گیرنده انسولین
گیرنده انسولین (انگلیسی: Insulin receptor) یک گیرندهٔ سطحی سلول است که با اتصال به انسولین، فاکتور رشد شبه انسولین ۱ و فاکتور رشد شبه انسولین ۲ فعال میشود و متعلق به خانوادهٔ بزرگ تیروزین کینازهای گیرندهای است.[۴]




گیرنده انسولین نقش بسیار مهمی در تنظیم قند خون ایفا میکند، فرآیندی عملکردی که تحت شرایط خاص ممکن است منجر به طیف وسیعی از تظاهرات بالینی از جمله دیابت و سرطان شود.[۵][۶] پیامدهی انسولین، دسترسی سلولهای بدن به گلوکز خون را کنترل میکند. هنگامی که انسولین کاهش مییابد، بهویژه در کسانی که حساسیت زیادی به انسولین دارند، سلولهای بدن شروع به انتقال و مصرف چربیهایی میکنند که نیازی به انتقال از طریق غشاء ندارند. به این ترتیب، انسولین تنظیمکننده مهمی در فرایند سوختوساز چربی نیز هست. از نظر بیوشیمیایی، گیرنده انسولین توسط یک ژن رمزگذاری میشود که «INSR» نام دارد و طی یک پیرایش دگرسان به یکی از دو ایزوفرم IR-A یا IR-B مبدل میشود.[۷] رویدادهای پاییندستی پس از ترجمهٔ هر یک از ایزوفرمها، منجر به تشکیل پروتئولیتیکی زیرواحدهای آلفا و بتا میشوند که پس از ترکیب، در نهایت قادر به هومو یا هترودایمرسازی برای تولید گیرنده انسولین تراغشایی ۳۲۰ کیلو دالتونی با پیوندهای دی سولفید هستند.[۷]
ساختار
ویرایشدر ابتدا، رونویسی پیرایشهای دگرسان مشتقشده از ژن «INSR» برای تشکیل یکی از دو ایزومر مونومریک ترجمه میشود: IR-A که در آن اگزون ۱۱ حذف شدهاست، و IR-B که در آن اگزون ۱۱ گنجانده شدهاست. گنجاندن اگزون ۱۱ منجر به افزودن ۱۲ اسید آمینه در جایگاهِ بالادستِ محل برش پروتئولیتیک فیورین درونزاد میشود.

بهدنبال دایمریزاسیون گیرنده، پس از برش آنزیمی زنجیرههای آلفا و بتا، ۱۲ اسید آمینه اضافی یادشده در پایانه-C زنجیرهٔ آلفا باقی میمانند (به آن αCT میگویند) و احتمالاً بر تعامل میان لیگاند و گیرنده اثر میگذارند. [۸]
هر مونومر ایزومری به لحاظ آرایش ساختاری، از ۸ دومِـین مجزا، یک دومِـین توالی سرشار از لوسین (L1، ریشههای ۱ تا ۱۵۷)، یک ناحیه غنی از سیستئین (CR، ریشههای ۱۵۸ تا ۳۱۰)، یک توالی سرشار از لوسین دیگر (L2، ریشه هیا ۳۱۱ تا ۴۷۰)، سه دومین فیبرونکتین نوع ۳ با نامهای «FnIII-1» (ریشههای ۴۷۱ تا ۵۹۵)، «FnIII-2» (ریشههای ۵۹۶ تا ۸۰۸) و «FnIII-3» (ریشههای ۸۰۹ تا ۹۰۶) سازماندهی شدهاست. علاوه بر اینها، یک دومِـین تونهاده (ID، ریشههای ۶۳۸ تا ۷۵۶) در درون دومِـین «FnIII-2» وجود دارد که حاوی جایگاه تجزیه و شکافت فیورین آلفا/بتا است که منجر به تشکیل دومِـینهای IDα و IDβ میگردد. در زنجیره بتای پاییندستِ دومِـین FnIII-3 یک مارپیچ تراغشایی (TH) و ناحیه کنارغشایی درونسلولی (JM) قرار دارد، درست در بالادستِ دومِـین کاتالیتیک درونسلولیِ تیروزین کیناز (TK) که مسئول مسیرهای پیامرسانی درونسلولی بعدی است.[۹]
پس از تفکیک مونومر به زنجیرههای آلفا و بتا مربوطهاش، هترو یا همودایمریزاسیون گیرنده انسولین به صورت کووالانسی میان زنجیرهها توسط یک پیوند دیسولفیدی (و میان مونومرهای یک مولکول دوتایی توسط دو پیوند دیسولفیدی) که از هر زنجیره آلفا بیرون زده، حفظ میشود. ساختار سه بعدی کلی اکتودومین که دارای چهار محل اتصال لیگاند است، شبیه یک "V" معکوس است، و هر مونومر تقریباً به اندازهٔ ۲ پیچ، حول محوری به موازات دومِـینهای "V" معکوس و L2 و FnIII-1 از هر مونومر چرخیدهاست و راس "V" را تشکیل میدهد.[۹][۱۰]
اتصال به لیگاند
ویرایش

لیگاندهای درونزای گیرنده انسولین شامل انسولین، فاکتور رشد شبه انسولین ۱ و فاکتور رشد شبه انسولین ۲ است. در سالهای اخیر، با استفاده از میکروسکوپ الکترونی کرایو، پژوهشگران به بینش ساختاری جدیدی نسبت به تغییرات ساختمانی این گیرنده پس از اتصال به انسولین دست یافتند. اتصال لیگاند به زنجیرههای α اکتودومین دایمر IR، آن را از یک V معکوس به یک ترکیب T شکل تغییر میدهد و این تغییر از نظر ساختاری به تمامی دومِـینهای غشایی تسری مییابد و در نهایت منجر به اتوفسفوریلاسیون ریشههای تیروزینی دومِـین تیروزین کیناز (TK) در زنجیرهٔ بتا میشود.[۱۱] این تغییرات جذب برخی پروتئینهای خاص وفقدهنده ترارسانی پیام را تسهیل میکند که از آن میان میتوان به پروتئینهای سوبسترای گیرندهٔ انسولین (IRS) , SHB (هومولوژی ۲ -بی C-Src)، مولکول APS و پروتئینفسفاتازهایی چون PTPN1 اشاره کرد. همه اینها در نهایت منجر به فرایندهای پائیندستی میشود که در همایستایی گلوکز و تنظیم قند خون نقش دارند.[۱۳]
بررسیهای دقیق نشان داده که رابطهٔ بین گیرنده انسولین و لیگاند مشخصات آلوستریک پیچیدهای دارد. این خواص پیچیده با استفاده از نمودارهای اسکاچارد اثبات شد که با استفاده از آن، مشخص گشت که نسبت «لیگاندِ متصل به گیرنده» به «لیگاند غیر متصل به گیرنده» از یک رابطه خطی (با توجه به تغییرات غلظت لیگاند متصل به گیرنده) برخوردار نیست و نشان میدهد که گیرنده انسولین و لیگاندش، رابطه ای از نوع پیوند برهمکنشی دارند.[۱۴] علاوه بر اینها، مشاهدهٔ اینکه سرعت تفکیک لیگاند از گیرنده انسولین با افزودن لیگاند آزاد (غیر متصل) تسریع میشود، نشان میدهد که این پیوند برهمکنشی ماهیتی منفی دارد. عبارت دیگر، اتصال اولیه لیگاند به گیرنده انسولین، جلوی اتصال بیشتر لیگاند به دومین جایگاه فعال گیرنده را میگیرد که نمودی از بازدارندگی آلوستریک است.[۱۴]
این مدلها بیان میکنند که هر مونومر گیرنده انسولین دارای ۲ جایگاه اتصال به انسولین است. جایگاه ۱، که به سطح اتصالی «کلاسیک» انسولین متصل میشود و متشکل از دومِـینهای L1 به اضافه αCT است و جایگاه ۲، متشکل از حلقههایی در محل اتصال دومِـینهای FnIII-1 و FnIII-2 که پیشبینی میشود به سطح اتصالی «نوین» انسولین (که آرایشی شش تایی دارد) متصل شود.[۴] از آنجایی که هر مونومر دخیل در اکتودومین گیرندهٔ انسولین، رفتار متمم «آینهای» سهبعدی از خود نشان میدهد، جایگاه ۱ پایانه-N در یک مونومر در نهایت با جایگاه ۲ پایانه-C در مونومر دوم روبروی هم قرار میگیرند، این نحوه آرایش برای هر مونومر مکمل آینهای نیز صدق میکند (در سمت مقابل ساختار اکتودومین). متون علمی کنونی، جایگاههای اتصال مکمل را با تغییر نامگذاری جایگاههای ۱ و ۲ در مونومر دوم بهترتیب به جایگاه ۳ و جایگاه ۴ یا به صورت جایگاه ۱' و جایگاه ۲' مشخص میکند.[۴][۱۳] به این ترتیب، این مدلها بیان میکنند که هر گیرندهٔ انسولین ممکن است از طریق ۴ جایگاه اتصالی به یک مولکول انسولین (که دارای دو سطح اتصال است) پیوند یابد، که جایگاههای ۱، ۲، (۳/۱') یا (۴/۲') است. مطابق با مدلسازی ریاضی فعلی از سینتیک (مکانیک) انسولین با گیرندهاش، دو پیامد مهم برای اتصال عرضی انسولین وجود دارد. (الف) که با مشاهدات فوقالذکر از پیوند برهمکنشی منفی میان گیرندهٔ انسولین و لیگاندش، اتصال بعدی لیگاند به گیرنده کاهش مییابد و (ب) عمل فیزیکی اتصال عرضی است که اکتودومین را به چنین ترکیبی میرساند و این ساختار فضایی برای فسفوریلاسیون تیروزین درونسلولی لازم است. (یعنی این واکنشها، از ملزومات مهم فعالشدنِ گیرنده و حفظ نهایی همایستایی قند خون است).[۱۳]
دانشمندان توانستند با استفاده از شبیهسازیهای میکروسکوپ الکترونی کرایو و دینامیک ملکولی گیرنده بازسازیشده در نانودیسکها، ساختار دایمری کل اکتودومین گیرنده انسولین را با چهار مولکول انسولین متصلشده به آن مشاهده کنند؛ بنابراین وجودِ ۴ جایگاه اتصالی پیشبینیشده توسط علم بیوشیمی تأیید و مستقیماً اثبات شد.[۱۲]
آگونیستها
ویرایشچندین مولکول کوچک آگونیست دیگر برای گیرنده انسولین کشف شدهاست.[۱۵]
مسیر ترارسانی و انتقال پیامهای سلولی-مولکولی
ویرایشگیرنده انسولین نوعی گیرنده تیروزین کیناز است که در آن اتصال یک لیگاند آگونیستی باعث اتوفسفوریلاسیون ریشههای تیروزین میشود و هر زیرواحد، جفت مولکولی خود را فسفریله میکند. افزودن گروههای فسفات یک جایگاه اتصالی برای سوبسترای گیرنده انسولین (IRS-1) ایجاد میکند که متعاقباً از طریق فسفوریلاسیون فعال میشود. سوبسترای فعالشده، مسیر انتقال پیامهای سلولی را آغاز میکند و به فسفواینوزیتید ۳-کیناز متصل میشود که به نوبه خود باعث فعال شدن این مولکول میگردد و تبدیل فسفاتیدیلاینوزیتول (۴٬۵) بیسفسفات به فسفاتیدیلاینوزیتول (۳٬۴٬۵) تریفسفات (PIP3) را تسهیل میکند. PIP3 به عنوان یک پیامرسان ثانویه عمل میکند و باعث فعال شدن پروتئین کیناز وابسته به فسفاتیدیلاینوزیتول میشود، که سپس چندین کیناز دیگر را فعال میکند - بهویژه پروتئین کیناز بی (PKB، که با عنوان Akt هم شناخته میشود). پروتئین کیناز بی، از طریق فعال سازی پروتئینهای SNARE، باعث انتقال وزیکولهای حاوی گلوکز (GLUT4) به غشای سلولی میشود تا انتشار گلوکز به داخل سلول را تسهیل کند. پروتئین کیناز بی همچنین گلیکوژن سنتاز کیناز ۳ را که آنزیم بازدارندهٔ گلیکوژن سنتاز است، فسفریله و مهار میکند؛ بنابراین، پروتئین کیناز بی در آغاز فرایند گلیکوژنز هم نقش دارد که در نهایت غلظت قند خون را کاهش میدهد.[۱۶]
-
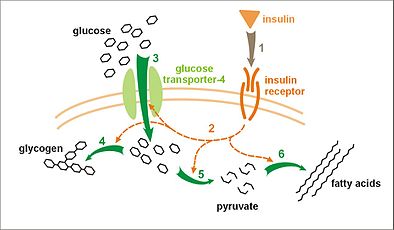 تأثیر انسولین بر جذب و سوختوساز گلوکز. انسولین به گیرنده خود متصل میشود (۱)، که به نوبه خود، آبشارهای فعالسازی بسیاری از پروتئین را آغاز میکند (۲). اینها عبارتند از: انتقال حامل شمارهٔ ۴ گلوکز به غشای پلاسمایی و درونریزی گلوکز (۳)، ساخت گلیکوژن (۴)، گلیکولیز (۵)، و ساخت اسیدهای چرب (۶).
تأثیر انسولین بر جذب و سوختوساز گلوکز. انسولین به گیرنده خود متصل میشود (۱)، که به نوبه خود، آبشارهای فعالسازی بسیاری از پروتئین را آغاز میکند (۲). اینها عبارتند از: انتقال حامل شمارهٔ ۴ گلوکز به غشای پلاسمایی و درونریزی گلوکز (۳)، ساخت گلیکوژن (۴)، گلیکولیز (۵)، و ساخت اسیدهای چرب (۶). -
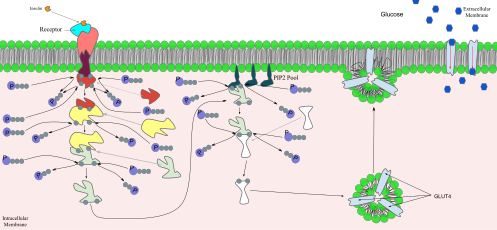 ترارسانی سلولی-مولکولی پیام انسولین: در پایان فرایند ترارسانی پیام، پروتئین فعالشده به پروتئینهای فسفاتیدیلاینوزیتول (۴٬۵) بیسفسفات تعبیهشده در جدارهٔ سلول متصل میشود.
ترارسانی سلولی-مولکولی پیام انسولین: در پایان فرایند ترارسانی پیام، پروتئین فعالشده به پروتئینهای فسفاتیدیلاینوزیتول (۴٬۵) بیسفسفات تعبیهشده در جدارهٔ سلول متصل میشود.


عملکرد
ویرایشتنظیم بیان ژن
ویرایشIRS-1 فعالشده به عنوان یک پیامرسان ثانویه در سلول عمل میکند تا رونویسی ژنهای تنظیمشونده با انسولین را تحریک کند. ابتدا، پروتئین شمارهٔ ۲ متصل به گیرندهٔ فاکتور رشد به ریشهٔ فسفوتیروزین IRS-1 در دومِـین SH2 خود متصل میشود. سپس GRB2 میتواند به ژن SOS متصل شود، که به نوبه خود موجب جایگزینی گوانوزین دیفسفات با گوانوزین تریفسفات در مولکول Ras میگردد که نوعی جیپروتئین است. سپس این پروتئین یک آبشار فسفریلاسیون را آغاز میکند و منجر به ایجاد یک پروتئین کیناز فعالشده با میتوژن میگردد که وارد هستهٔ سلول شده و عوامل مختلف رونویسی درونهستهای (همچون پروتئین ELK1) را فسفریله میکند.
تحریک ساخت گلیکوژن
ویرایشساخت گلیکوژن نیز توسط گیرنده انسولین از طریق IRS-1 تحریک میشود. در این حالت، دومین SH2 فسفواینوزیتید ۳-کیناز است که به ریشهٔ فسفوتیروزین IRS-1 متصل میشود. فسفواینوزیتید ۳-کیناز فعالشده، فسفاتیدیلاینوزیتول (۴٬۵) بیسفسفات غشایی را به فسفاتیدیلاینوزیتول (۳٬۴٬۵) تریفسفات تبدیل میکند. این کار بهطور غیر مستقیم پروتئین کیناز بی را از طریق فسفریلاسیون فعال میکند. پروتئین کیناز بی به نوبهٔ خود چندین پروتئین دیگر، از جمله گلیکوژن سنتاز کیناز ۳ را تحریک میکند. گلیکوژن سنتاز کیناز ۳، مسئول فسفریلاسیون و غیرفعالسازی آنزیم گلیکوژن سنتاز است. وقتی گلیکوژن سنتاز کیناز ۳ فسفریله میشود، غیرفعال میگردد و بدین ترتیب آنزیم گلیکوژن سنتاز هم فعال نمیشود. به این ترتیب انسولین سنتز گلیکوژن را افزایش میدهد.
تجزیهٔ انسولین
ویرایشهنگامی که یک مولکول انسولین به گیرندهاش متصل شد و وظیفهاش را انجام داد، یا دوباره به محیط برونسلولی رها میشود یا توسط سلول تجزیه میشود. مراحل تجزیه معمولاً شامل درونبری ترکیب گیرنده-انسولین و به دنبال آن کنشِ آنزیم تجزیهکننده انسولین است. بیشتر مولکولهای انسولین توسط سلولهای کبد تجزیه میشوند. تخمین زده شدهاست که یک مولکول معمولی انسولین در نهایت حدود ۷۱ دقیقه پس از انتشار اولیه آن در گردش خون تجزیه میگردد.[۱۷]
دستگاه ایمنی
ویرایشعلاوه بر عملکرد سوختوسازی، گیرندههای انسولین بر روی سلولهای دستگاه ایمنی مانند ماکروفاژها، لنفوسیتهای بی و لنفوسیتهای تی نیز بیان میشوند. گیرندههای انسولین در لنفوسیتهای تی در حالت استراحت چندان دیده نمیشوند، اما با فعال شدن گیرنده لنفوسیت تی (TCR) بیان آن افزایش مییابد. در واقع، ثابت شدهاست که زمانی که انسولین به صورت اگزوژن (برونزاد یا دارویی) در شرایط آزمایشگاهی به مدلهای حیوانی عرضه میشود، تکثیر لنفوسیتهای تی را در آنان تقویت میکند. سیگنالدهی گیرنده انسولین برای به حداکثر رساندن اثر بالقوه لنفوسیتهای تی در طول عفونت حاد و التهاب حائز اهمیت است.[۱۸][۱۹]
ارتباط با بیماریها
ویرایشوظیفهٔ اصلی گیرنده انسولین فعالشده، افزایش جذب گلوکز توسط سلولهاست. به همین دلیل «عدم حساسیت به انسولین» یا کاهش پیامرسانی گیرنده انسولین منجر به دیابت نوع ۲ میشود: سلولها قادر به جذب گلوکز نیستند و نتیجه هایپرگلیسمی (افزایش قند خون در گردش) و تمام عواقب بالقوهٔ ناشی از این بیماری ممکن است رخ دهد.
مبتلایان به مقاومت به انسولین گاهی علامت پوستی آکانتوز نیگریکانس دارند.
تاکنون چند بیمار با جهش هموزیگوت در ژن INSR شرح داده شدهاست که باعث سندرم داناهیو یا «لپریکانیسم» میشود. در این اختلال ژنتیکی اتوزومال مغلوب گیرنده انسولین کاملاً فاقد عملکرد طبیعی خود است. این بیماران دارای گوشهای کوچک برجسته، اغلب، سوراخهای بینی باز، لبهای کلفت و دچار عقبافتادگی شدید رشد هستند. در بیشتر موارد پیشآگهی این بیماری در نوزادان مبتلا بسیار بد است و در سال اول زندگی میمیرند. جهشهای دیگر همان ژن باعث اختلال خفیفتری به نام سندرم رابسون-مندنهال میشود که در آن بیماران دارای دندانهای غیرطبیعی خاص، لثههای هیپرتروفیک و بزرگ شدن غده پینهآل هستند. در هر دو بیماری نوسانات قند خون وجود دارد: بعد از غذا، گلوکز در ابتدا بسیار بالا است و سپس به سرعت به سطوح غیرطبیعی افت میکند. سایر جهشهای ژنتیکی در ژن گیرنده انسولین میتوانند باعث مقاومت شدید به انسولین شوند.[۲۰] Other genetic mutations to the insulin receptor gene can cause Severe Insulin Resistance.[۲۱]
تعاملهای شیمیایی
ویرایشگیرنده انسولین با مولکولهای زیر تعامل پروتئین-پروتئین دارد.
منابع
ویرایش- ↑ ۱٫۰ ۱٫۱ ۱٫۲ GRCm38: Ensembl release 89: ENSMUSG00000005534 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ ۴٫۰ ۴٫۱ ۴٫۲ Ward CW, Lawrence MC (April 2009). "Ligand-induced activation of the insulin receptor: a multi-step process involving structural changes in both the ligand and the receptor". BioEssays. 31 (4): 422–34. doi:10.1002/bies.200800210. PMID 19274663. S2CID 27645596.
- ↑ Ebina Y, Ellis L, Jarnagin K, Edery M, Graf L, Clauser E, Ou JH, Masiarz F, Kan YW, Goldfine ID (April 1985). "The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling". Cell. 40 (4): 747–58. doi:10.1016/0092-8674(85)90334-4. PMID 2859121. S2CID 23230348.
- ↑ Malaguarnera R, Sacco A, Voci C, Pandini G, Vigneri R, Belfiore A (May 2012). "Proinsulin binds with high affinity the insulin receptor isoform A and predominantly activates the mitogenic pathway". Endocrinology. 153 (5): 2152–63. doi:10.1210/en.2011-1843. PMID 22355074.
- ↑ ۷٫۰ ۷٫۱ Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R (October 2009). "Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease". Endocrine Reviews. 30 (6): 586–623. doi:10.1210/er.2008-0047. PMID 19752219.
- ↑ Knudsen L, De Meyts P, Kiselyov VV (December 2011). "Insight into the molecular basis for the kinetic differences between the two insulin receptor isoforms" (PDF). The Biochemical Journal. 440 (3): 397–403. doi:10.1042/BJ20110550. PMID 21838706.
- ↑ ۹٫۰ ۹٫۱ Smith BJ, Huang K, Kong G, Chan SJ, Nakagawa S, Menting JG, Hu SQ, Whittaker J, Steiner DF, Katsoyannis PG, Ward CW, Weiss MA, Lawrence MC (April 2010). "Structural resolution of a tandem hormone-binding element in the insulin receptor and its implications for design of peptide agonists". Proceedings of the National Academy of Sciences of the United States of America. 107 (15): 6771–6. Bibcode:2010PNAS..107.6771S. doi:10.1073/pnas.1001813107. PMC 2872410. PMID 20348418.
- ↑ McKern NM, Lawrence MC, Streltsov VA, Lou MZ, Adams TE, Lovrecz GO, Elleman TC, Richards KM, Bentley JD, Pilling PA, Hoyne PA, Cartledge KA, Pham TM, Lewis JL, Sankovich SE, Stoichevska V, Da Silva E, Robinson CP, Frenkel MJ, Sparrow LG, Fernley RT, Epa VC, Ward CW (September 2006). "Structure of the insulin receptor ectodomain reveals a folded-over conformation". Nature. 443 (7108): 218–21. Bibcode:2006Natur.443..218M. doi:10.1038/nature05106. PMID 16957736. S2CID 4381431.
- ↑ ۱۱٫۰ ۱۱٫۱ Gutmann T, Kim KH, Grzybek M, Walz T, Coskun Ü (May 2018). "Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor". The Journal of Cell Biology. 217 (5): 1643–1649. doi:10.1083/jcb.201711047. PMC 5940312. PMID 29453311.
- ↑ ۱۲٫۰ ۱۲٫۱ Gutmann T, Schäfer IB, Poojari C, Brankatschk B, Vattulainen I, Strauss M, Coskun Ü (January 2020). "Cryo-EM structure of the complete and ligand-saturated insulin receptor ectodomain". The Journal of Cell Biology. 219 (1). doi:10.1083/jcb.201907210. PMC 7039211. PMID 31727777.
- ↑ ۱۳٫۰ ۱۳٫۱ ۱۳٫۲ Kiselyov VV, Versteyhe S, Gauguin L, De Meyts P (Feb 2009). "Harmonic oscillator model of the insulin and IGF1 receptors' allosteric binding and activation". Molecular Systems Biology. 5 (5): 243. doi:10.1038/msb.2008.78. PMC 2657531. PMID 19225456.
- ↑ ۱۴٫۰ ۱۴٫۱ de Meyts P, Roth J, Neville DM, Gavin JR, Lesniak MA (November 1973). "Insulin interactions with its receptors: experimental evidence for negative cooperativity". Biochemical and Biophysical Research Communications. 55 (1): 154–61. doi:10.1016/S0006-291X(73)80072-5. PMID 4361269.
- ↑ Kumar L, Vizgaudis W, Klein-Seetharaman J (July 2022). "Structure-based survey of ligand binding in the human insulin receptor". Br J Pharmacol. 179 (14): 3512–3528. doi:10.1111/bph.15777. PMID 34907529. S2CID 245242018.
- ↑ Berg JM, Tymoczko J, Stryer L, Berg JM, Tymoczko JL, Stryer L (2002). Biochemistry (5th ed.). W H Freeman. ISBN 0716730510.
- ↑ Duckworth WC, Bennett RG, Hamel FG (October 1998). "Insulin degradation: progress and potential". Endocrine Reviews. 19 (5): 608–24. doi:10.1210/edrv.19.5.0349. PMID 9793760.
- ↑ Tsai S, Clemente-Casares X, Zhou AC, Lei H, Ahn JJ, Chan YT, et al. (August 2018). "Insulin Receptor-Mediated Stimulation Boosts T Cell Immunity during Inflammation and Infection". Cell Metabolism. 28 (6): 922–934.e4. doi:10.1016/j.cmet.2018.08.003. PMID 30174303.
- ↑ Fischer HJ, Sie C, Schumann E, Witte AK, Dressel R, van den Brandt J, Reichardt HM (March 2017). "The Insulin Receptor Plays a Critical Role in T Cell Function and Adaptive Immunity". Journal of Immunology. 198 (5): 1910–1920. doi:10.4049/jimmunol.1601011. PMID 28115529.
- ↑ Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D (June 2002). "Genotype-phenotype correlation in inherited severe insulin resistance". Human Molecular Genetics. 11 (12): 1465–75. doi:10.1093/hmg/11.12.1465. PMID 12023989. S2CID 15924838.
- ↑ Melvin A, Stears A (2017). "Severe insulin resistance: pathologies". Practical Diabetes (به انگلیسی). 34 (6): 189–194a. doi:10.1002/pdi.2116. S2CID 90238599. Retrieved 2020-10-31.
- ↑ Maddux BA, Goldfine ID (January 2000). "Membrane glycoprotein PC-1 inhibition of insulin receptor function occurs via direct interaction with the receptor alpha-subunit". Diabetes. 49 (1): 13–9. doi:10.2337/diabetes.49.1.13. PMID 10615944.
- ↑ Langlais P, Dong LQ, Hu D, Liu F (June 2000). "Identification of Grb10 as a direct substrate for members of the Src tyrosine kinase family". Oncogene. 19 (25): 2895–903. doi:10.1038/sj.onc.1203616. PMID 10871840.
- ↑ Hansen H, Svensson U, Zhu J, Laviola L, Giorgino F, Wolf G, Smith RJ, Riedel H (April 1996). "Interaction between the Grb10 SH2 domain and the insulin receptor carboxyl terminus". The Journal of Biological Chemistry. 271 (15): 8882–6. doi:10.1074/jbc.271.15.8882. PMID 8621530.
- ↑ Liu F, Roth RA (October 1995). "Grb-IR: a SH2-domain-containing protein that binds to the insulin receptor and inhibits its function". Proceedings of the National Academy of Sciences of the United States of America. 92 (22): 10287–91. Bibcode:1995PNAS...9210287L. doi:10.1073/pnas.92.22.10287. PMC 40781. PMID 7479769.
- ↑ He W, Rose DW, Olefsky JM, Gustafson TA (March 1998). "Grb10 interacts differentially with the insulin receptor, insulin-like growth factor I receptor, and epidermal growth factor receptor via the Grb10 Src homology 2 (SH2) domain and a second novel domain located between the pleckstrin homology and SH2 domains". The Journal of Biological Chemistry. 273 (12): 6860–7. doi:10.1074/jbc.273.12.6860. PMID 9506989.
- ↑ Frantz JD, Giorgetti-Peraldi S, Ottinger EA, Shoelson SE (January 1997). "Human GRB-IRbeta/GRB10. Splice variants of an insulin and growth factor receptor-binding protein with PH and SH2 domains". The Journal of Biological Chemistry. 272 (5): 2659–67. doi:10.1074/jbc.272.5.2659. PMID 9006901.
- ↑ Kasus-Jacobi A, Béréziat V, Perdereau D, Girard J, Burnol AF (April 2000). "Evidence for an interaction between the insulin receptor and Grb7. A role for two of its binding domains, PIR and SH2". Oncogene. 19 (16): 2052–9. doi:10.1038/sj.onc.1203469. PMID 10803466.
- ↑ Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF (January 2002). "Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action". The Journal of Biological Chemistry. 277 (2): 1531–7. doi:10.1074/jbc.M101521200. PMID 11606564.
- ↑ Sawka-Verhelle D, Tartare-Deckert S, White MF, Van Obberghen E (March 1996). "Insulin receptor substrate-2 binds to the insulin receptor through its phosphotyrosine-binding domain and through a newly identified domain comprising amino acids 591-786". The Journal of Biological Chemistry. 271 (11): 5980–3. doi:10.1074/jbc.271.11.5980. PMID 8626379.
- ↑ O'Neill TJ, Zhu Y, Gustafson TA (April 1997). "Interaction of MAD2 with the carboxyl terminus of the insulin receptor but not with the IGFIR. Evidence for release from the insulin receptor after activation". The Journal of Biological Chemistry. 272 (15): 10035–40. doi:10.1074/jbc.272.15.10035. PMID 9092546.
- ↑ Braiman L, Alt A, Kuroki T, Ohba M, Bak A, Tennenbaum T, Sampson SR (April 2001). "Insulin induces specific interaction between insulin receptor and protein kinase C delta in primary cultured skeletal muscle". Molecular Endocrinology. 15 (4): 565–74. doi:10.1210/mend.15.4.0612. PMID 11266508.
- ↑ Rosenzweig T, Braiman L, Bak A, Alt A, Kuroki T, Sampson SR (June 2002). "Differential effects of tumor necrosis factor-alpha on protein kinase C isoforms alpha and delta mediate inhibition of insulin receptor signaling". Diabetes. 51 (6): 1921–30. doi:10.2337/diabetes.51.6.1921. PMID 12031982.
- ↑ Maegawa H, Ugi S, Adachi M, Hinoda Y, Kikkawa R, Yachi A, Shigeta Y, Kashiwagi A (March 1994). "Insulin receptor kinase phosphorylates protein tyrosine phosphatase containing Src homology 2 regions and modulates its PTPase activity in vitro". Biochemical and Biophysical Research Communications. 199 (2): 780–5. doi:10.1006/bbrc.1994.1297. PMID 8135823.
- ↑ Kharitonenkov A, Schnekenburger J, Chen Z, Knyazev P, Ali S, Zwick E, White M, Ullrich A (December 1995). "Adapter function of protein-tyrosine phosphatase 1D in insulin receptor/insulin receptor substrate-1 interaction". The Journal of Biological Chemistry. 270 (49): 29189–93. doi:10.1074/jbc.270.49.29189. PMID 7493946.
- ↑ Kotani K, Wilden P, Pillay TS (October 1998). "SH2-Balpha is an insulin-receptor adapter protein and substrate that interacts with the activation loop of the insulin-receptor kinase". The Biochemical Journal. 335 (1): 103–9. doi:10.1042/bj3350103. PMC 1219757. PMID 9742218.
- ↑ Nelms K, O'Neill TJ, Li S, Hubbard SR, Gustafson TA, Paul WE (December 1999). "Alternative splicing, gene localization, and binding of SH2-B to the insulin receptor kinase domain". Mammalian Genome. 10 (12): 1160–7. doi:10.1007/s003359901183. PMID 10594240. S2CID 21060861.
برای مطالعهٔ بیشتر
ویرایش- Pearson RB, Kemp BE (1991). "Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations". Methods in Enzymology. 200: 62–81. doi:10.1016/0076-6879(91)00127-I. ISBN 978-0-12-182101-2. PMID 1956339.
- Joost HG (February 1995). "Structural and functional heterogeneity of insulin receptors". Cellular Signalling. 7 (2): 85–91. doi:10.1016/0898-6568(94)00071-I. PMID 7794689.
- O'Dell SD, Day IN (July 1998). "Insulin-like growth factor II (IGF-II)". The International Journal of Biochemistry & Cell Biology. 30 (7): 767–71. doi:10.1016/S1357-2725(98)00048-X. PMID 9722981.
- Lopaczynski W (1999). "Differential regulation of signaling pathways for insulin and insulin-like growth factor I". Acta Biochimica Polonica. 46 (1): 51–60. doi:10.18388/abp.1999_4183. PMID 10453981.
- Sasaoka T, Kobayashi M (August 2000). "The functional significance of Shc in insulin signaling as a substrate of the insulin receptor". Endocrine Journal. 47 (4): 373–81. doi:10.1507/endocrj.47.373. PMID 11075717.
- Perz M, Torlińska T (2001). "Insulin receptor--structural and functional characteristics". Medical Science Monitor. 7 (1): 169–77. PMID 11208515.
- Benaim G, Villalobo A (August 2002). "Phosphorylation of calmodulin. Functional implications". European Journal of Biochemistry. 269 (15): 3619–31. doi:10.1046/j.1432-1033.2002.03038.x. hdl:10261/79981. PMID 12153558.
پیوند به بیرون
ویرایش- Insulin receptor در سرعنوانهای موضوعی پزشکی (MeSH) در کتابخانهٔ ملی پزشکی ایالات متحدهٔ آمریکا
- خلاصهای از اطلاعات ساختاری موجود در بانک داده پروتئین برای یونیپروت: P06213 (Insulin receptor) در PDBe-KB.